

ニチアスは 「断つ・保つ」®の技術で
地球の明るい未来に貢献します
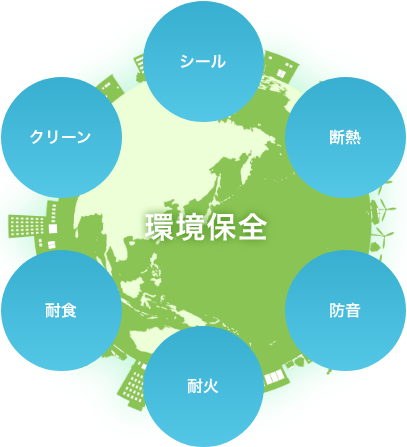
ニチアスは、「シール」「断熱」「防音」「耐火」「耐食」「クリーン」といった「断つ・保つ」®の技術を基盤とした5つの事業でさまざまな産業分野を支え、地球環境の保全に貢献しております。
各種プラント設備向けに製品やエンジニアリングを提供する「プラント向け工事・販売事業」をはじめ、基幹産業を主な市場とする「工業製品事業」、半導体産業に特化した「高機能製品事業」、自動車メーカーなどを主な客先とする「自動車部品事業」、ビルや住宅の建材を供給・施工する「建材事業」、いずれの事業もこれまでに培ってきた技術や経験を活かしながら、グローバル化を推進しています。





製品案内
Products Information
お知らせ
News
- すべて
- お知らせ
- 製品関連
- イベント
- WEBセミナー講演のお知らせ【Thermofit® (省エネ診断システム)~隠れた熱ロスの発見とその対策~】
- 展示会「INCHEM TOKYO 2023」に出展します
- 展示会「BATTERY JAPAN【秋】(二次電池展)」に出展します
IR ニュース
IR News
会社案内
Company




サステナビリティ
Sustainability







お問い合わせ
Contact

.JPG)

